Harmonising datasets
harmonise.RmdRun
Harmonise two datasets. Ensure that both GWAS inputs are standardised
with standardise_gwas() and that the appropriate columns to
join on are specified in the merge argument - usually
either c("CHR"="CHR","BP"="BP") or
c("RSID"="RSID").
library(genepi.utils)
# the gwas data
gwas1 <- GWAS(dat=system.file("extdata", "example2_gwas_sumstats.tsv", package="genepi.utils"), map="ns_map")
#> Loading GWAS ...
#> [i] loading data from: example2_gwas_sumstats.tsv
#> [i] applying column mapping
#> [i] enforcing column types
#> [i] standardising columns
#> [i] standardising allele coding
#> [i] applying filters
#> [i] checking rsid coding
#> [?] 10 rsids could not be parsed, setting to chr:bp
gwas2 <- GWAS(dat=system.file("extdata", "example2_gwas_sumstats.tsv", package="genepi.utils"), map="ns_map")
#> Loading GWAS ...
#> [i] loading data from: example2_gwas_sumstats.tsv
#> [i] applying column mapping
#> [i] enforcing column types
#> [i] standardising columns
#> [i] standardising allele coding
#> [i] applying filters
#> [i] checking rsid coding
#> [?] 10 rsids could not be parsed, setting to chr:bp
gwas1 <- as.data.table(gwas1)
gwas2 <- as.data.table(gwas2)
# mess with some alleles
gwas1[c(5,7,8), c("ea","oa") := list(c("T","I","I"),c("A","D","D"))]
gwas2[c(5,6,7,8), c("ea","oa") := list(c("A","A","D","ACC"),c("T","A","I","A"))]
# view
gwas2
#> rsid chr bp ea oa eaf beta se p
#> <char> <char> <int> <char> <char> <num> <num> <num> <num>
#> 1: 10:100000625 10 100000625 G A 7.10e-05 -0.037 0.137 0.781
#> 2: 10:100000645 10 100000645 C A 1.21e-01 0.045 0.173 0.961
#> 3: 10:100003242 10 100003242 G T 5.04e-07 -0.037 0.134 0.480
#> 4: 10:100003785 10 100003785 C T 1.55e-01 -0.008 0.137 0.743
#> 5: 10:100004360 10 100004360 A T 8.38e-01 0.045 0.134 0.961
#> 6: 10:100004906 10 100004906 A A 7.40e-01 -0.037 0.173 0.781
#> 7: 10:100004996 10 100004996 D I 3.23e-10 -0.008 0.137 0.743
#> 8: 10:100005282 10 100005282 ACC A 9.30e-04 -0.147 0.134 0.781
#> 9: 10:100007362 10 100007362 C G 8.16e-05 0.045 0.173 0.961
#> 10: 10:100008436 10 100008436 A G 9.46e-04 -0.008 0.208 0.743
#> n ncase trait id strand imputed info q q_p i2
#> <int> <int> <char> <char> <char> <lgcl> <num> <num> <num> <num>
#> 1: NA NA trait id <NA> NA NA 0.781 NA NA
#> 2: NA NA trait id <NA> NA NA 0.961 NA NA
#> 3: NA NA trait id <NA> NA NA 0.480 NA NA
#> 4: NA NA trait id <NA> NA NA 0.743 NA NA
#> 5: NA NA trait id <NA> NA NA 0.961 NA NA
#> 6: NA NA trait id <NA> NA NA 0.781 NA NA
#> 7: NA NA trait id <NA> NA NA 0.743 NA NA
#> 8: NA NA trait id <NA> NA NA 0.781 NA NA
#> 9: NA NA trait id <NA> NA NA 0.961 NA NA
#> 10: NA NA trait id <NA> NA NA 0.743 NA NA
# harmonise
harmonised <- harmonise(gwas1, gwas2, merge=c("chr"="chr","bp"="bp"))[]
# view
harmonised
#> Key: <chr_incidence, bp_incidence>
#> rsid_incidence rsid_progression chr_incidence chr_progression bp_incidence
#> <char> <char> <char> <char> <int>
#> 1: 10:100000625 10:100000625 10 10 100000625
#> 2: 10:100000645 10:100000645 10 10 100000645
#> 3: 10:100003242 10:100003242 10 10 100003242
#> 4: 10:100003785 10:100003785 10 10 100003785
#> 5: 10:100004360 10:100004360 10 10 100004360
#> 6: 10:100004906 10:100004906 10 10 100004906
#> 7: 10:100004996 10:100004996 10 10 100004996
#> 8: 10:100005282 10:100005282 10 10 100005282
#> 9: 10:100007362 10:100007362 10 10 100007362
#> 10: 10:100008436 10:100008436 10 10 100008436
#> bp_progression ea_incidence ea_progression oa_incidence oa_progression
#> <int> <char> <char> <char> <char>
#> 1: 100000625 G G A A
#> 2: 100000645 C C A A
#> 3: 100003242 G G T T
#> 4: 100003785 C C T T
#> 5: 100004360 T T A A
#> 6: 100004906 A A C A
#> 7: 100004996 I I D D
#> 8: 100005282 ACC ACC A A
#> 9: 100007362 C C G G
#> 10: 100008436 A A G G
#> eaf_incidence eaf_progression beta_incidence beta_progression p_incidence
#> <num> <num> <num> <num> <num>
#> 1: 7.10e-05 7.10e-05 -0.037 -0.037 0.781
#> 2: 1.21e-01 1.21e-01 0.045 0.045 0.961
#> 3: 5.04e-07 5.04e-07 -0.037 -0.037 0.480
#> 4: 1.55e-01 1.55e-01 -0.008 -0.008 0.743
#> 5: 8.38e-01 1.62e-01 0.045 -0.045 0.961
#> 6: 7.40e-01 7.40e-01 -0.037 -0.037 0.781
#> 7: 3.23e-10 1.00e+00 -0.008 0.008 0.743
#> 8: 9.30e-04 9.30e-04 -0.147 -0.147 0.781
#> 9: 8.16e-05 8.16e-05 0.045 0.045 0.961
#> 10: 9.46e-04 9.46e-04 -0.008 -0.008 0.743
#> p_progression palindromic keep se_incidence n_incidence ncase_incidence
#> <num> <lgcl> <lgcl> <num> <int> <int>
#> 1: 0.781 FALSE TRUE 0.137 NA NA
#> 2: 0.961 FALSE TRUE 0.173 NA NA
#> 3: 0.480 FALSE TRUE 0.134 NA NA
#> 4: 0.743 FALSE TRUE 0.137 NA NA
#> 5: 0.961 TRUE TRUE 0.134 NA NA
#> 6: 0.781 FALSE FALSE 0.173 NA NA
#> 7: 0.743 FALSE TRUE 0.137 NA NA
#> 8: 0.781 FALSE TRUE 0.134 NA NA
#> 9: 0.961 TRUE TRUE 0.173 NA NA
#> 10: 0.743 FALSE TRUE 0.208 NA NA
#> trait_incidence id_incidence strand_incidence imputed_incidence
#> <char> <char> <char> <lgcl>
#> 1: trait id <NA> NA
#> 2: trait id <NA> NA
#> 3: trait id <NA> NA
#> 4: trait id <NA> NA
#> 5: trait id <NA> NA
#> 6: trait id <NA> NA
#> 7: trait id <NA> NA
#> 8: trait id <NA> NA
#> 9: trait id <NA> NA
#> 10: trait id <NA> NA
#> info_incidence q_incidence q_p_incidence i2_incidence se_progression
#> <num> <num> <num> <num> <num>
#> 1: NA 0.781 NA NA 0.137
#> 2: NA 0.961 NA NA 0.173
#> 3: NA 0.480 NA NA 0.134
#> 4: NA 0.743 NA NA 0.137
#> 5: NA 0.961 NA NA 0.134
#> 6: NA 0.781 NA NA 0.173
#> 7: NA 0.743 NA NA 0.137
#> 8: NA 0.781 NA NA 0.134
#> 9: NA 0.961 NA NA 0.173
#> 10: NA 0.743 NA NA 0.208
#> n_progression ncase_progression trait_progression id_progression
#> <int> <int> <char> <char>
#> 1: NA NA trait id
#> 2: NA NA trait id
#> 3: NA NA trait id
#> 4: NA NA trait id
#> 5: NA NA trait id
#> 6: NA NA trait id
#> 7: NA NA trait id
#> 8: NA NA trait id
#> 9: NA NA trait id
#> 10: NA NA trait id
#> strand_progression imputed_progression info_progression q_progression
#> <char> <lgcl> <num> <num>
#> 1: <NA> NA NA 0.781
#> 2: <NA> NA NA 0.961
#> 3: <NA> NA NA 0.480
#> 4: <NA> NA NA 0.743
#> 5: <NA> NA NA 0.961
#> 6: <NA> NA NA 0.781
#> 7: <NA> NA NA 0.743
#> 8: <NA> NA NA 0.781
#> 9: <NA> NA NA 0.961
#> 10: <NA> NA NA 0.743
#> q_p_progression i2_progression
#> <num> <num>
#> 1: NA NA
#> 2: NA NA
#> 3: NA NA
#> 4: NA NA
#> 5: NA NA
#> 6: NA NA
#> 7: NA NA
#> 8: NA NA
#> 9: NA NA
#> 10: NA NAEvaluation speed
The number of variants for evaluation speed analysis was downsampled to 100,000, as otherwise it takes a very long time.
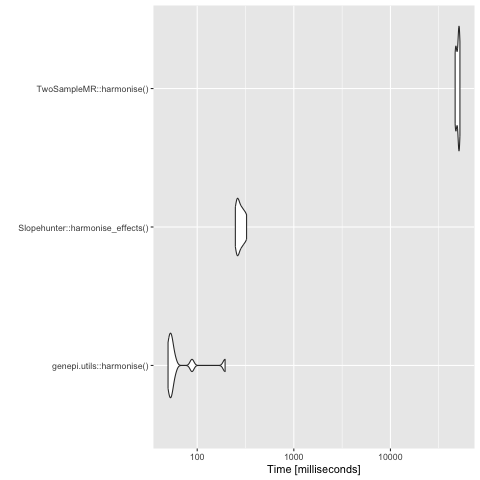
Analysis with downsampled GWAS of 100,000 variants
library(microbenchmark)
library(ggplot2)
gwas_progression_path <- "/Users/xx20081/Documents/local_data/hermes_progression/biostat_disc/post_qc/biostat_disc.allcause_death.autosomes.gz"
gwas_incidence_path <- "/Users/xx20081/Documents/local_data/hermes_incidence/standardised/hf_incidence_pheno1_eur.tsv.gz"
# some GWAS data
gwas1 <- data.table::fread(gwas_incidence_path)[1:100000, ]
gwas2 <- gwas1
# Slopehunter read in
gwas1_sh <- SlopeHunter::read_incidence(gwas_incidence_path, snp_col="SNP", beta_col="BETA", se_col="SE", pval_col="P", eaf_col="EAF", effect_allele_col="EA", other_allele_col="OA", chr_col = "CHR", pos_col="POS")[1:100000, ]
gwas2_sh <- SlopeHunter::read_prognosis(gwas_incidence_path, snp_col="SNP", beta_col="BETA", se_col="SE", pval_col="P", eaf_col="EAF", effect_allele_col="EA", other_allele_col="OA", chr_col = "CHR", pos_col="POS")[1:100000, ]
# TwoSampleMR read in
gwas1_2smr <- TwoSampleMR::format_data(gwas1 |> as.data.frame(), type="exposure", snp_col="SNP", beta_col="BETA", se_col="SE", pval_col="P", eaf_col="EAF", effect_allele_col="EA", other_allele_col="OA", chr_col = "CHR", pos_col="POS")[1:100000, ]
gwas2_2smr <- TwoSampleMR::format_data(gwas1 |> as.data.frame(), type="outcome", snp_col="SNP", beta_col="BETA", se_col="SE", pval_col="P", eaf_col="EAF", effect_allele_col="EA", other_allele_col="OA", chr_col = "CHR", pos_col="POS")[1:100000, ]
# benchmarking harmonisation
mbm <- microbenchmark("genepi.utils::harmonise()" = { genepi.utils::harmonise(gwas1, gwas2, merge=c("CHR"="CHR","BP"="BP")) },
"Slopehunter::harmonise_effects()" = { SlopeHunter::harmonise_effects(gwas1, gwas2) },
"TwoSampleMR::harmonise()" = { TwoSampleMR::harmonise_data(gwas1_2smr, gwas2_2smr) },
times = 10L)
# 2SMR issue fixed in recent commit
# mbm <- microbenchmark("TwoSampleMR::harmonise() - updated" = { TwoSampleMR::harmonise_data(gwas1_2smr, gwas2_2smr) },
# times = 10L)
autoplot(mbm)
# save
png("figures/microbenchmark_harmonise.png")
print(autoplot(mbm))
dev.off()